
Hi,
I am trying to see interaction of glycolic acid calcium salt on the tri calcium silicate (C3S) crystal surface using Interface-Pcff force field.
I imported C3S minimized unit cell from Interface force field documents. Then cleaved it and made 0 thickness vacuum slab. Then fixed the all position of atoms.
Then I created calcium glycolate molecule and optimized using DMOL and assigned Mullikan charges for the molecule.
Then I optimized the molecule using Interface-Pcff with current charge option. Then I created a confined layer of calcium glycolate and water. (Same a=b parameter as the C3S surface and experimental density)
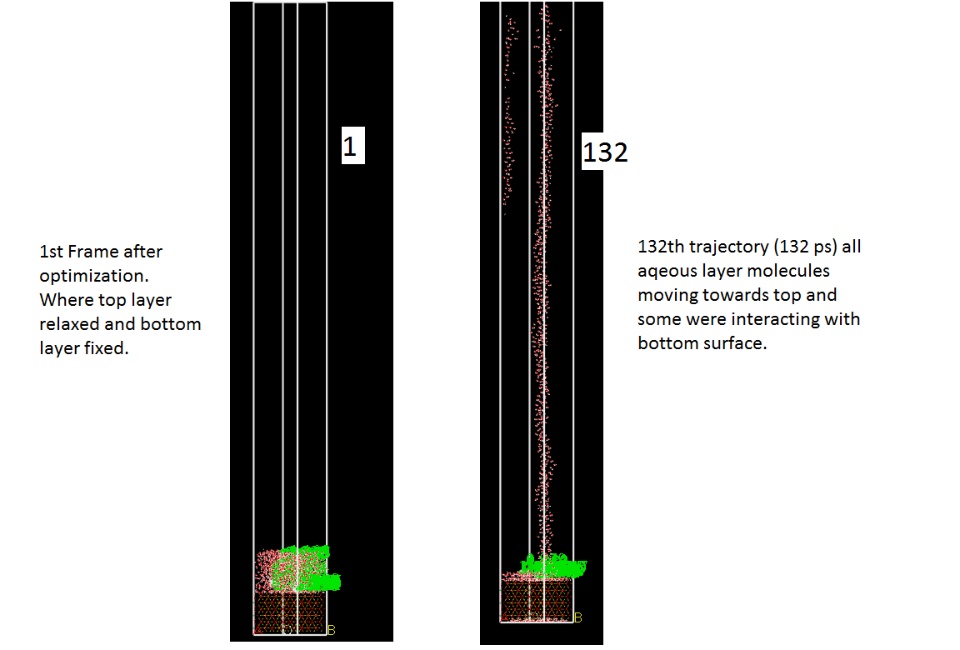
By using build layer, I created Calcium glycolate -water and C3S layer. (Shown in the image 1). I gave vacuum for the second layer 300 A, (when I created 60 and 150 A vacuum layers after geometry optimization molecules moved like a stream of water towards the periodic cell roof so I added 300 A vacuum). I cross checked the total charge which was zero.
So the box size became 37X35X351 A. Then I optimized the box using Interface-Pcff force field (current charges, auto force filed OFF). After that I ran the dynamics for 1000 ps , NVT-NOSE. Interface –pcff force field.
I found that after ~100 ps energy got decrease and all molecules from aqueous layer were moving towards the roof also they were start interacting with the bottom of the surface. (Image 2).
This is the first time I am observing this problem. I saw this problem during geometry optimization when I added 60 A and 150 A vacuum, but I thought it’s due to less vacuum so I added 300 A vacuum.
But then during dynamics I observed the same problem again.
How I can resolve the problem? Should I add more vacuum (more than 300A)?
Thank you for the help.
I am trying to see interaction of glycolic acid calcium salt on the tri calcium silicate (C3S) crystal surface using Interface-Pcff force field.
I imported C3S minimized unit cell from Interface force field documents. Then cleaved it and made 0 thickness vacuum slab. Then fixed the all position of atoms.
Then I created calcium glycolate molecule and optimized using DMOL and assigned Mullikan charges for the molecule.
Then I optimized the molecule using Interface-Pcff with current charge option. Then I created a confined layer of calcium glycolate and water. (Same a=b parameter as the C3S surface and experimental density)
By using build layer, I created Calcium glycolate -water and C3S layer. (Shown in the image 1). I gave vacuum for the second layer 300 A, (when I created 60 and 150 A vacuum layers after geometry optimization molecules moved like a stream of water towards the periodic cell roof so I added 300 A vacuum). I cross checked the total charge which was zero.
So the box size became 37X35X351 A. Then I optimized the box using Interface-Pcff force field (current charges, auto force filed OFF). After that I ran the dynamics for 1000 ps , NVT-NOSE. Interface –pcff force field.
I found that after ~100 ps energy got decrease and all molecules from aqueous layer were moving towards the roof also they were start interacting with the bottom of the surface. (Image 2).
This is the first time I am observing this problem. I saw this problem during geometry optimization when I added 60 A and 150 A vacuum, but I thought it’s due to less vacuum so I added 300 A vacuum.
But then during dynamics I observed the same problem again.
How I can resolve the problem? Should I add more vacuum (more than 300A)?
Thank you for the help.
