
Hi all,
I am trying to use the Geometry Optimization in Forcite Calculation on a monomer, but some atoms will go random as following.
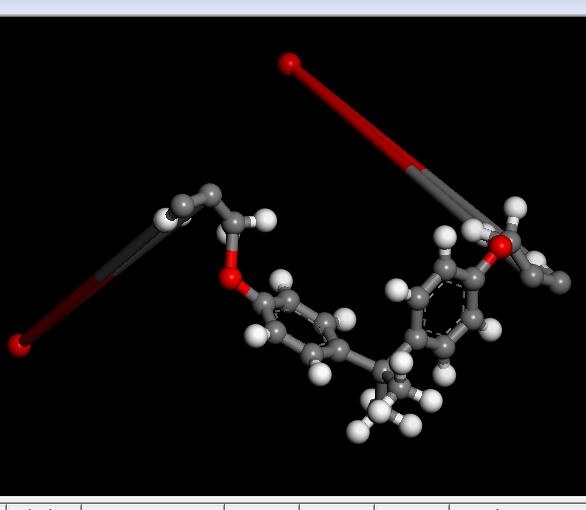
I have no idea about this situation. Is there any setting for the bond?
Best regards,
Anthea
I am trying to use the Geometry Optimization in Forcite Calculation on a monomer, but some atoms will go random as following.
I have no idea about this situation. Is there any setting for the bond?
Best regards,
Anthea
